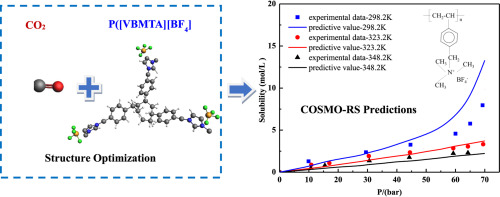
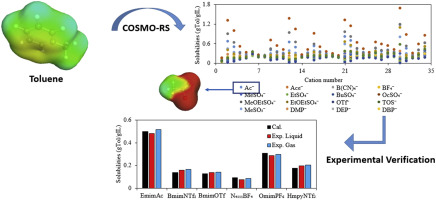
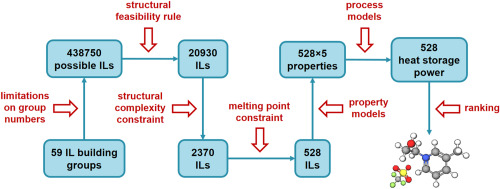
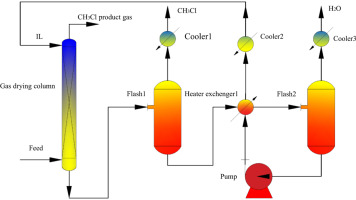
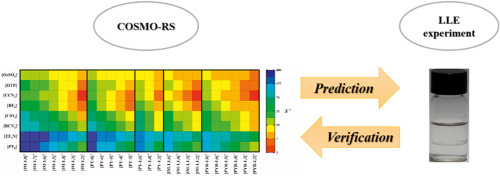
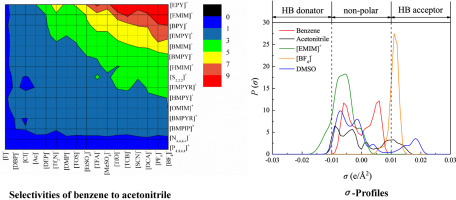
Rational design of ionic liquids (ILs), which is highly dependent on the accuracy of the model used, has always been crucial for CO2 separation from flue gas. In this study, a support vector machine (SVM) model which is a machine learning approach is established, so as to improve the prediction accuracy and range of IL melting points. Based on IL melting points data with 600 training data and 168 testing data, the estimated average absolute relative deviations (AARD) and squared correlation coefficients (R2) are 3.11%, 0.8820 and 5.12%, 0.8542 for the training set and testing set of the SVM model, respectively. Then, through the melting points model and other rational design processes including conductor-like screening model for real solvents (COSMO-RS) calculation and physical property constraints, cyano-based ILs are obtained, in which tetracyanoborate [TCB]- is often ruled out due to incorrect estimation of melting points model in the literature. Subsequently, by means of process simulation using Aspen Plus, optimal IL are compared with excellent IL reported in the literature. Finally, 1-ethyl-3-methylimidazolium tricyanomethanide [EMIM][TCM] is selected as a most suitable solvent for CO2 separation from flue gas, the process of which leads to 12.9% savings on total annualized cost compared to that of 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)amide [EMIM][Tf2N].








 京公网安备 11010802039050号
京公网安备 11010802039050号